Pipeline
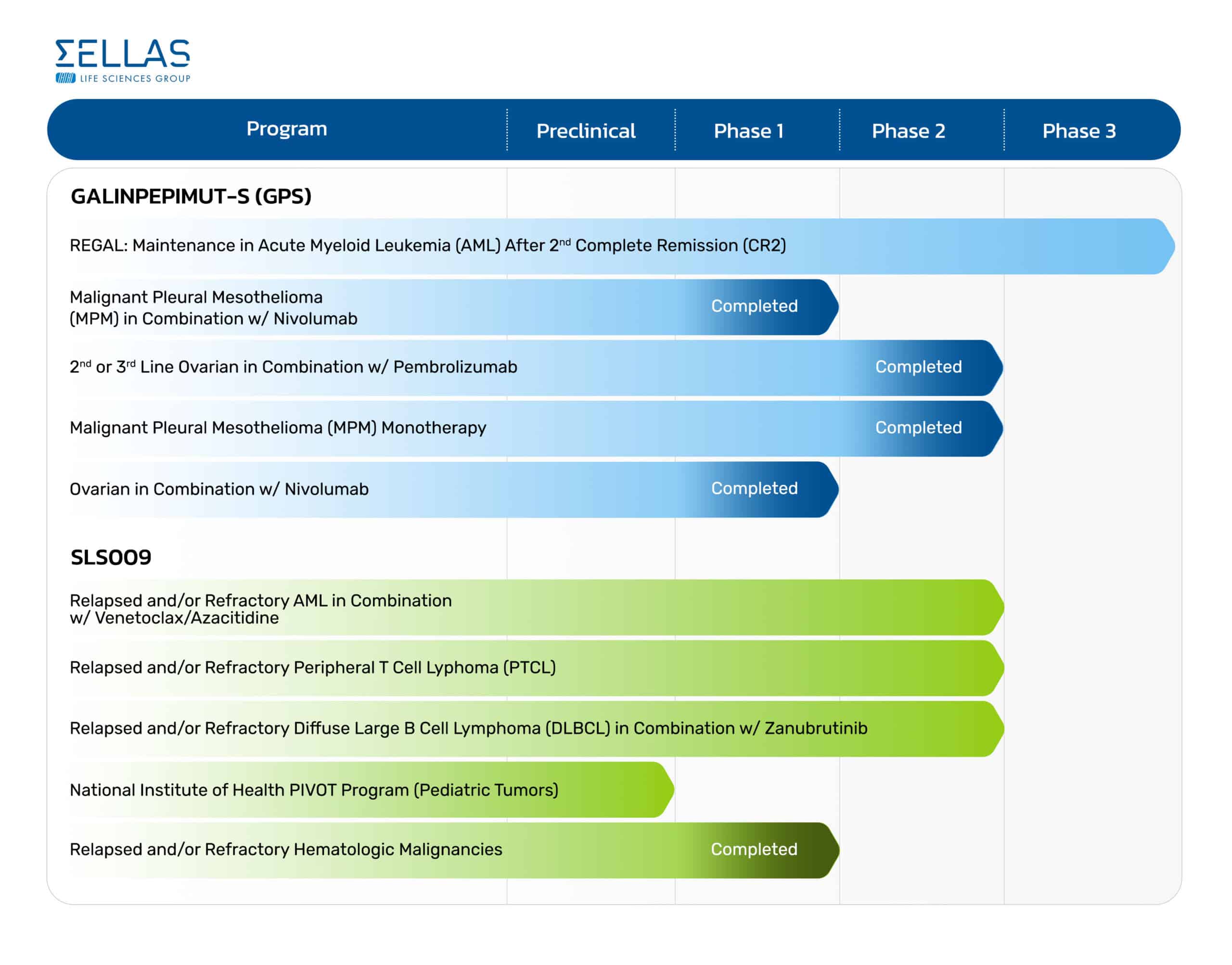
Galinpepimut-S (GPS)
Galinpepimut-S (GPS) is an immunotherapeutic agent licensed from Memorial Sloan Kettering Cancer Center, or MSK, that targets the Wilms Tumor 1 (WT1) protein which is present and over expressed in 20 or more cancer types and ranked by the National Cancer Institute (NCI) as the top priority among cancer antigens for immunotherapy. Traditional approaches have not yet been proven to successfully address the WT1 protein, as WT1 is not druggable with small molecules and is intracellular and inaccessible to antibodies. In patients with advanced cancers over-expressing WT1, GPS is designed to prevent relapses and potentially improve survival rates of patients, while maintaining their quality of life. Based on its mechanism of action as a directly immunizing agent, GPS has potential as a monotherapy or in combination with other immunotherapeutic agents to address a broad spectrum of hematologic, or blood, cancers and solid tumor indications.
SLS009
In March 2022, we entered into an exclusive license agreement with GenFleet Therapeutics, a clinical-stage biotechnology developing cutting-edge therapeutics in oncology and immunology, that grants rights to us for the development and commercialization of SLS009, a highly selective small molecule CDK9 inhibitor, across all therapeutic and diagnostic uses worldwide outside of the Greater China Region.
Indications
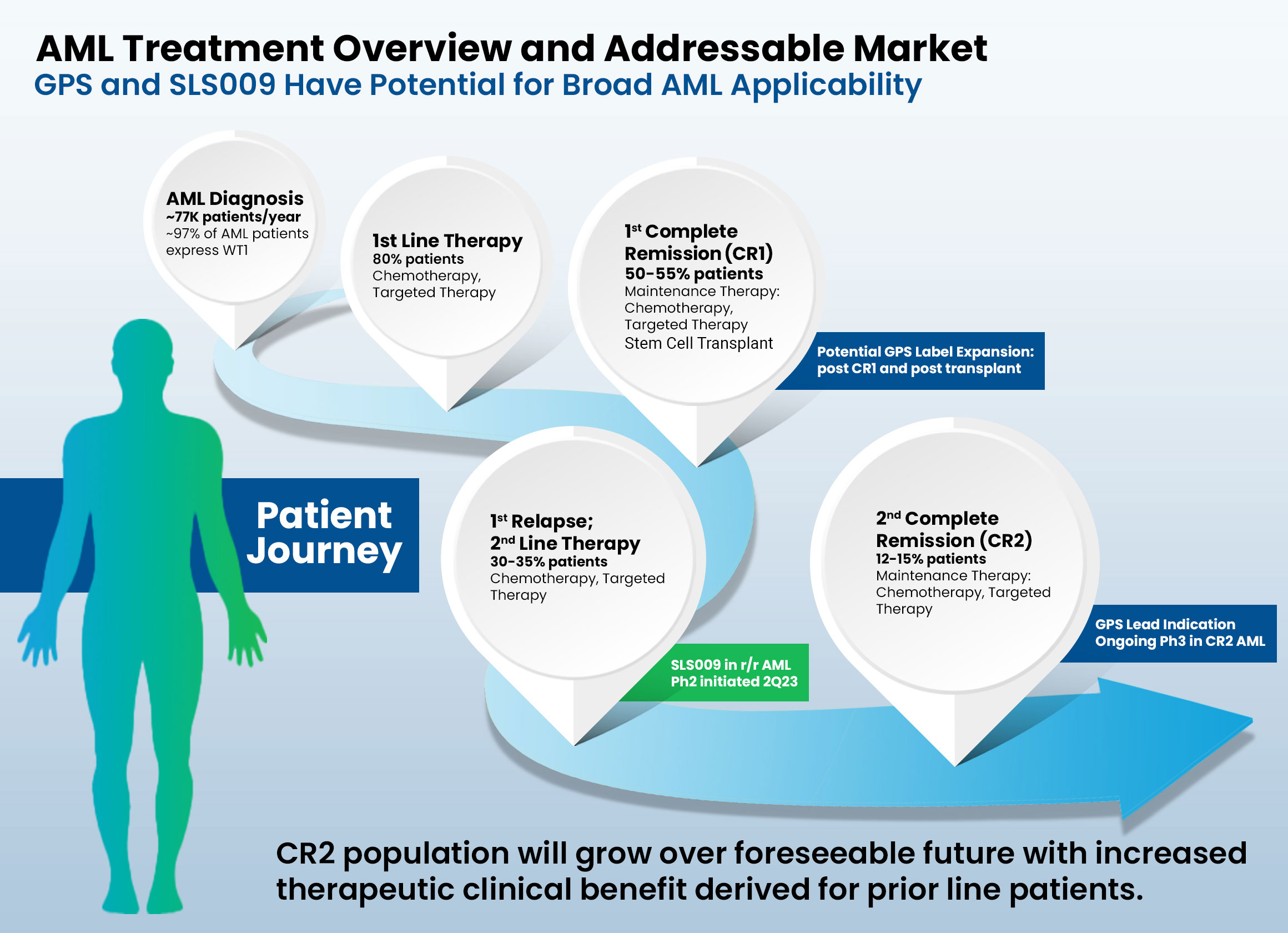
Acute Myeloid Leukemia (AML)
- Most commonly affects adults and incidence increases with age.
- Characterized by rapid growth of abnormal white blood cells that build up in the bone marrow and interfere with the production of normal blood cells.
- Cause of AML is unknown in most cases and is typically fatal within weeks or months if untreated.
- To date, other than an allogeneic hematopoietic stem cell transplant (HSCT), no therapies have been proven to be curative after patients achieve a complete response (CR) status. Once the disease relapses, then ‘second-line’ therapies can be given, but they have very limited positive clinical impact.
- A June 2021 report from Delvelnsight estimates a global market size for AML of $5.09 billion by the end of 2030, with a compound annual growth rate (CAGR) of 21.85% from 2018 to 2030.
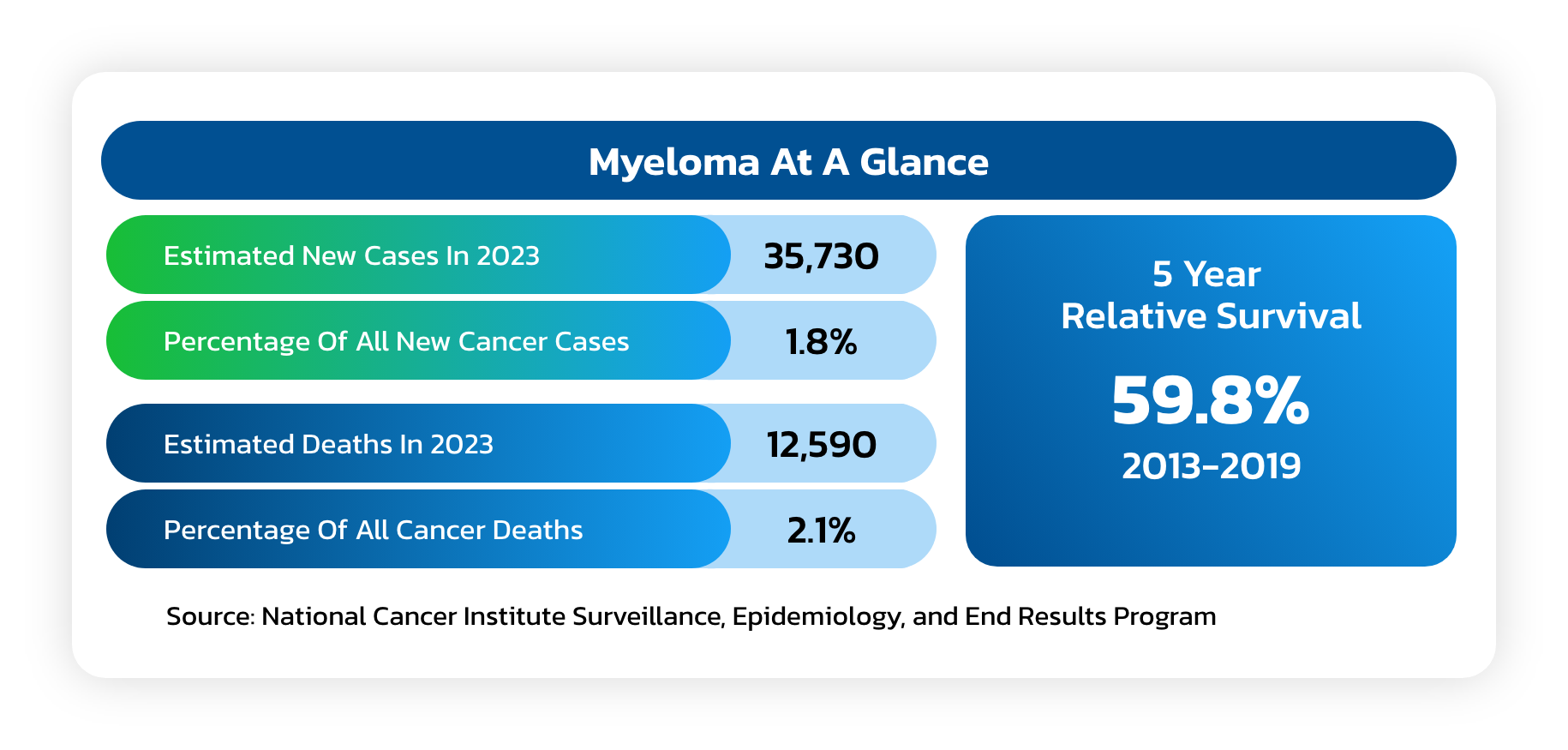
Multiple Myeloma
- A cancer formed by malignant plasma cells, and its cause is unknown.
- Overgrowth of plasma cells in the bone marrow crowds out normal blood-forming cells, causing low blood counts and anemia (a shortage of red blood cells) which can lead to increased bleeding and bruising, along with problems fighting infections.
- Causes a host of organ problems and symptoms, including fatigue, bone pain, fractures and kidney failure.
- Median survival can vary from up to at least 15 years in non-high-risk patients who achieve complete remission (CR) to about 3 years (from time of initial treatment) in patients with MM who achieve less than partial response (PR) after allogeneic stem cell transplant (ASCT).
- Despite significant therapeutic advances in the management of MM, the prognosis of patients with high risk cytogenetics at the time of diagnosis remains quite poor, even when they successfully complete an allogeneic stem cell transplant (ASCT), especially if they continue to have evidence of minimal residual disease (MRD).
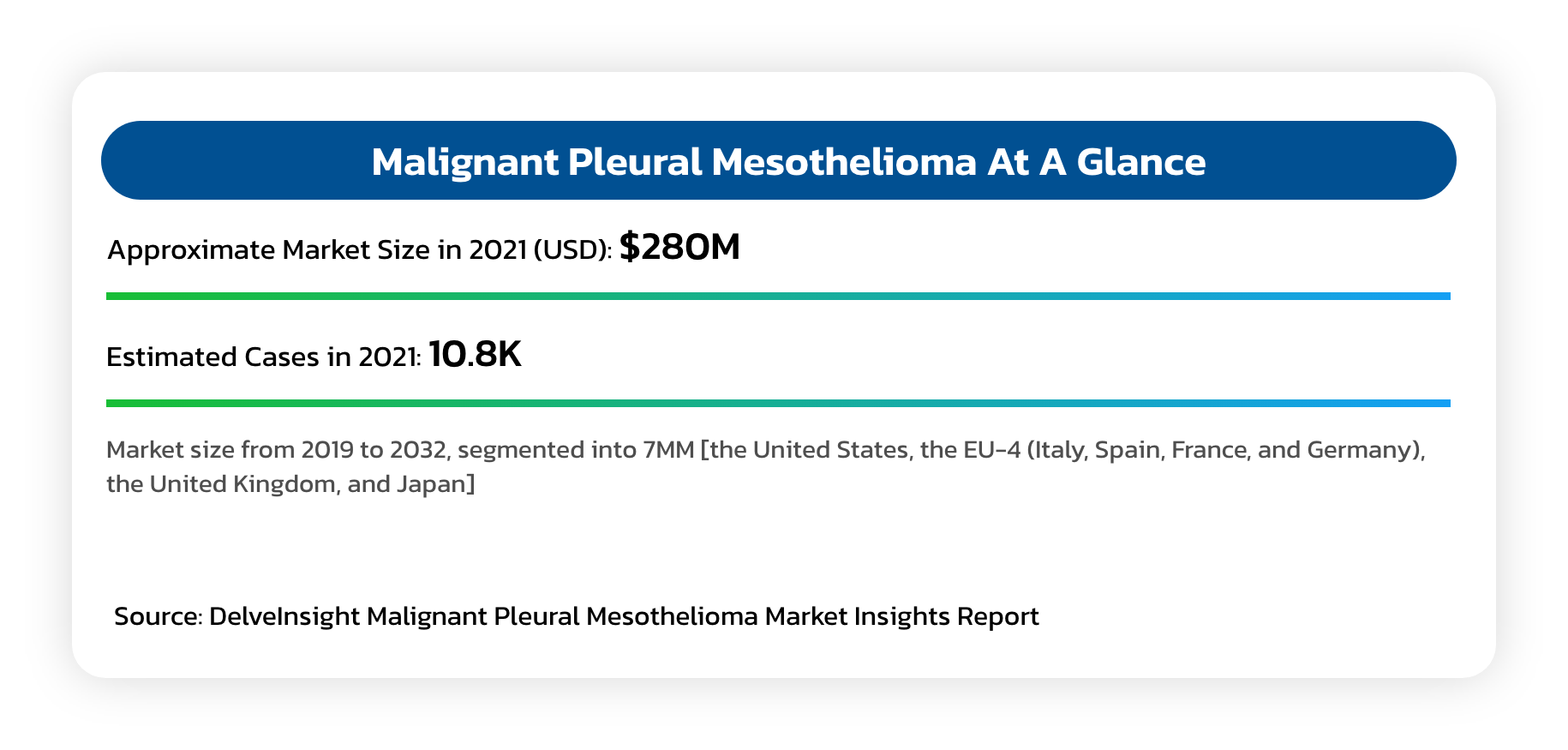
Malignant Pleural Mesothelioma (MPM)
- An asbestos related cancer that forms on the protective tissues that cover lungs.
- Most cases are traced to job-related exposures to asbestos and can take around 40 years between exposure and cancer formation.
- Typical survival is between 12 and 16 months, although select patients who complete a full course of indicated chemotherapy can survive up to 21 months.
- Mesothelioma is generally resistant to radiation and chemotherapy, while long term survival is rare, even in cases where aggressive upfront debulking multimodality therapies are used.
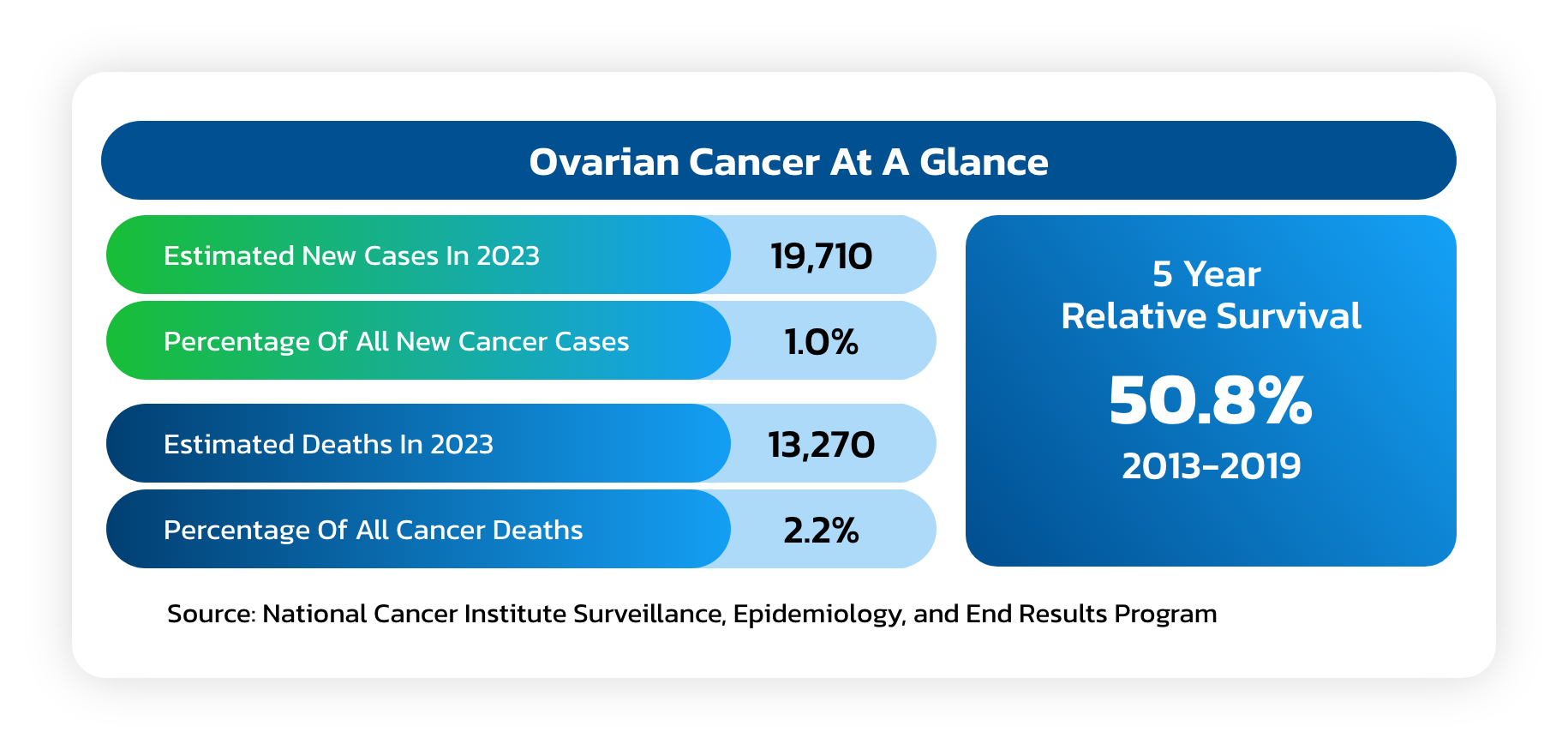
Ovarian Cancer
- A solid tumor that forms in the ovaries that ranks fifth in cancer deaths among women.
- Due to the lack of specific symptoms, the majority of ovarian cancer patients are
diagnosed at later stages of the disease, with an estimated 75% of women presenting
with advanced-stage (III or IV) disease. - A woman’s risk of getting ovarian cancer in her lifetime is about 1 in 78 and the lifetime
chance of dying from ovarian cancer is about 1 in 108. - The American Cancer Society estimates that in 2023 about 19,710 will receive a new
diagnosis of ovarian cancer and about 13,270 women will die from ovarian cancer.
Peripheral T-Cell Lymphoma (PTCL)
- PTCLs are uncommon and aggressive types of non-Hodgkin lymphoma (NHL) that develop in T cells and natural killer (NK) cells.
- 10-15% of all patients with NHL have a T-cell lymphoma subtype.
- PTCLs generally affect people older than 60 years although they can occur anytime during adulthood.
Patients: For more information about our clinical studies, please contact us at